

sanya
sanya@photonics.ru
Supramolecular approach means that you should deal with the dimer AB rather than with individual monomers A and B.
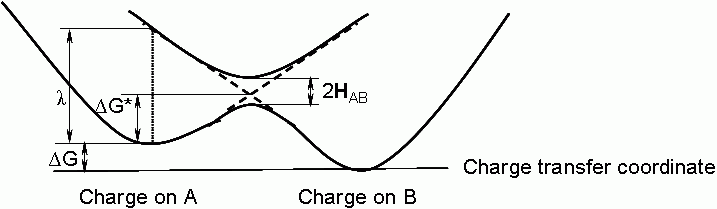
Strictly speaking, the charge-transfer integral in the Marcus model is the state splitting in the midpoint on the way from the state with the charge localized on A monomer to the state with the charge localized on B monomer (see picture). Therefore, the most adequate method is to use CASSCF for the entire energy profile from one minimum to the other. DFT seems to delocalize the charge equally between both monomers and, therefore, should not be used. After CASSCF, improve the energies by XMCQDPT. As a starting point, you can optimize the geometries of both minima (with charge localized on A and on B) by ROHF. Probably, you'll have to add dispersion correction to keep monomers together.
An alternative (although rather inaccurate and sometimes ambiguous) method is Energy Splitting in Dimer. Web of Science gives a lot of references on the use of this method. In short, you can take the splitting between HOMO and HOMO-1 (for hole hopping) or LUMO and LUMO-1 (for electron hopping) in the dimer. However, this method does not always work.
On Thu Mar 27 '14 9:54pm, pejer wrote
-------------------------------------
>Dear firefly user,
>in order to find charge transfer rate (Marcus-type expression) between two monomers, I have to find effective electronic coupling matrix element from that dimer. Manual said that, to define system with two monomers, I have to use supermolecular approach: calculate energy for each monomer, calculate energy for each monomer with dummy atoms of another monomer, etc. I didn't use Morokuma because I work with DFT. From this fact, I see that we couldn't define dimer system in one calculation. So, is there any probability to find effective electronic coupling matrix element? Is there someone who work out something like this too?
>I would really appreciate for any help.
>Best regards
Marcus picture
[ This message was edited on Sun Mar 30 '14 at 8:05pm by the author ]